
| Home | Documents | Reference | |||
| |||
 MethodsSNP functional analysisFor each pathway associated with trait, we firstly select the most significant SNPs of the significant genes (the genes mapped by at least one of the top 5 % SNPs). Then, we extract their linkage disequilibrium (LD) proxies (the SNPs having r2 > 0.8 with them) based on the HapMap III populations [1] or 1000 Genome populations [2]. For all the above SNPs, three types of annotation were performed: 1. ENCODE regulatory feature peaks [3]: five types of ENCODE Uniform Peaks were used to annotate SNPs into related regulatory regions, including DNase-seq peaks of open chromatin, FAIRE peaks of open chromatin, TFBS SPP-based peaks, TFBS PeakSeq-based peaks and Histone peaks. Detailed cell line and other information for each type of peaks is in here. 2. Putative functional information (from Ensembl [4]): annotated functional types included deleterious or probably/possibly damaging, splice donor variant, stop lost, incomplete terminal codon variant, inframe insertion, transcript ablation, splice acceptor variant, frameshift variant, stop gained, initiator codon variant, splice region variant or inframe deletion. Deleterious or probably/possibly damaging variants were denoted as "Deterious", others were denoted as "Others". 3. eQTL: SNP related eQTL were from several eQTL databases or browsers, including eQTL Browser, GTEx and seeQTL. The detailed data source description for the data included for eQTL analysis is in here. Finally we performed an enrichment test based on binomial distribution [5] to test if the SNPs are significantly enriched in functional elements compared with all Ensembl SNPs as background. To do this test, we first estimate empirical p as proportion of functional elements in the whole genome or all SNPs, depending on the functional element type. Then for m out of n SNPs are in functional elements, P-value is calculated as: 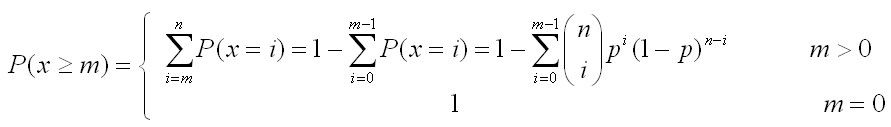 (1) (1)The P-values are calculated for each type of functional elements respectively, including each cell line of DNase-seq peaks (total 125) and FAIRE peaks (total 25), each TFBS cluster (total 495 for PeakSeq and SPP respectively), each Histone cluster (total 190), deleterious and others variants defined as above, as well as eQTL. Finally the P-values are corrected for number of cell lines, TFBS clusters or Histone clusters. i-GSEAThe i-GSEA4GWAS web server implements i-GSEA (improved gene set enrichment analysis) to help researchers explore GWAS data efficiently. i-GSEA is an implementation and extension of the original GSEA for GWAS. The key steps of i-GSEA are the same as GSEA with two highlights: 1) i-GSEA implements SNP label permutation instead of phenotype label permutation to adapt GWAS SNP P-values and to correct gene and gene set variation; 2) i-GSEA multiplies a significance proportion ratio factor to the ES to get the significant proportion based enrichment score (SPES) as described in details below. Briefly, firstly following the classical GSEA for gene expression study [6] and GSEA for GWAS [7], the maximum -log(P-value) or statistics of all the SNPs mapped to a gene was used to represent the gene (t). Then for N genes presented in GWAS, we ranked the genes by decreasing t(1) , t(2) ,..., t(i) ,..., t(N). For each given gene set S with set size Ns, the enrichment score, ES(S), with parameter w = 1 is calculated: 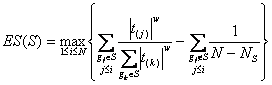 (2) (2)ES(S) emphasizes on the added-up significance of the top genes in S. High ES(S) indicates the association signal in S is highly concentrated at the top of the ranked gene list. Then the key step is: a significant cutoff t0 for the genes mapped with at least one of the top 5% of all SNPs is considered. Instead of ES(S), a significance proportion based enrichment score, SPES(S), is expressed as: 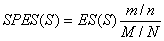 (3) (3)Where m is the number of genes in gene set S, n is the number of all genes in gene set S; M is the number of genes with t > t0 in the GWAS and N is the number of all the genes in the GWAS. SPES emphasizes on the proportion of significant genes in gene set S to avoid the high scoring caused by very few genes with extremely high significance. The following steps, variant label permutation, normalization, calculating gene set P value and FDR, are the same as the classical GSEA for GWAS [7]. Our previous work [8] has shown that i-GSEA has improved sensitivity in comparison to GSEA.
References |
|
Copyright: Bioinformatics Lab, Institute of Psychology, Chinese Academy of Sciences
Feedback Last update: April 18, 2014 |